Acta Biochimica Polonica, Vol. 65, No 1/2018, 59–66, https://doi.org/10.18388/abp.2017_1499
Regular paper
Cloning, expression and characterization of thermostable YdaP from Bacillus licheniformis 9A
Joseph D. Wani Lako1, Jada P. Yengkopiong1*, William H. L. Stafford2, Marla Tuffin3 and
Don A. Cowan4
1John Garang Memorial University of Science and Technology, College of Science and Technology, Bor, Jonglei State, Republic of South Sudan; 2CSIR Bioscience, Stellenbosch, Cape Town, South Africa; 3Institute for Microbial Biotechnology and Metagenomics (IMBM), Department of Biotechnology, University of the Western Cape, Bellville 7535, Cape Town, South Africa; 4Center of Genomics, University of Pretoria, Pretoria, South Africa
The Bacillus licheniformis ydaP gene encodes for a pyruvate oxidase that catalyses the oxidative decarboxylation of pyruvate to acetate and CO2. The YdaP form of this enzyme was purified about 48.6-folds to homogeneity in three steps. The enzyme was recovered in a soluble form and demonstrated significant activity on pyruvate using 2, 6-dichlorophenolindophenol (DCPIP) as an artificial electron acceptor. HPLC analysis of the YdaP-enzyme catalysed conversion of pyruvate showed acetate as the sole product, confirming the putative identity of pyruvate oxidase. Analysis of the substrate specificity showed that the YdaP enzyme demonstrated preference for short chain oxo acids; however, it was activated by 1% Triton X-100. The YdaP substrate-binding pocket from the YdaP protein differed substantially from the equivalent site in all of the so far characterized pyruvate oxidases, suggesting that the B. licheniformis YdaP might accept different substrates. This could allow more accessibility of large substrates into the active site of this enzyme. The thermostability and pH activity of the YdaP enzyme were determined, with optimums at 50ºC and pH 5.8, respectively. The amino acid residues forming the catalytic cavity were identified as Gln460 to Ala480.
Key words: Bacillus licheniformis, pyruvate oxidase, YdaP, thiamine diphosphate enzyme
Received: 06 January, 2017; revised: 09 April, 2017; accepted:
11 April, 2017; available on-line: 15 March, 2018
*e-mail: jadalojuka@yahoo.com.au
Abbreviations: POX, pyruvate oxidase, ThDP, thiamine diphosphate enzyme
Introduction
Thermophilic bacteria are a group of microorganisms that can grow and survive under broad environmental conditions, ranging from thermal pools to desert soils (Steele et al., 2008). This wide range of conditions indicates that the thermophilic bacteria produce enzymes that are stable and active under these extreme conditions. As a result, thermophilic bacteria are of interest to biotechnological and industrial applications because they work best at elevated temperatures.
Bacillus licheniformis is a thermophilic rod-shaped Gram-positive, facultative anaerobic bacterium, ubiquitous in nature and predominates soils (Claus & Berkeley, 1986). It grows optimally at around 55°C. B. licheniformis has been widely used in biotechnology and industry to manufacture enzymes, including α-amylase, cycloglucosyltransferase, β-mannanase, pectinolytic enzymes, penicillinase, pentosanase, proteases, antibiotics, biochemicals and other products (Eveleigh, 1981; Erickson, 1976). It is also used for microbial fermentations. As such, it is known as the ‘industrial’ bacterium. B. licheniformis genome has been sequenced (Rey et al., 2004), and it facilitates the production of other products with potential industrial uses.
The pyruvate oxidases (POX) are peripheral membrane associated flavoproteins belonging to the family of thiamine diphosphate dependent enzymes (Chang & Cronan, 1995). They are studied due to their importance in biotechnological applications. This includes the production of acetate for the food and beverage industries, and as biocatalysts for the production of acetylphosphate and other chemical intermediates (Yoneda et al., 2001; Tomar et al., 2003; Yilmaztekin et al., 2008). POX enzymes are present in most bacteria, and those from Escherichia coli (Mather et al., 1982), Corynebacterium glutamicum (Schreiner & Eikmanns, 2005), Staphylococcus aureus (Patton et al., 2005), Aerococcus viridans (Juan et al., 2007) and Lactobacillus plantarum (Sedewitz et al., 1984; Goffin et al., 2006; Lorquet et al., 2004) have been well characterized. These enzymes are divided into two subfamilies (Bertaggnolli & Hager, 1991). Subfamily I (EC 1.2.3.3), catalyzes the oxidative decarboxylation of pyruvate in the presence of oxygen and inorganic phosphate, generating hydrogen peroxide and acetylphosphate. Subfamily II (EC 1.2.5.1) catalyzes the oxidative decarboxylation of pyruvate, generating acetate and carbon dioxide in the presence of ferricytochrome b1 as an electron acceptor (Blake et al., 1982). These previously characterized pyruvate oxidases were shown to have activity at moderate temperatures (Sedewitz et al., 1984). Pyruvate oxidases are strongly activated by low concentrations of phospholipids and detergents (Chang & Cronan, 1986; Hamilton, 1986; Schreiner & Eikmanns, 2005). The phospholipid activation occurs by an association with the protein and is accompanied by conformational changes and alteration of various properties of the enzyme (Wang et al., 1991).
The objective of this study was to biochemically characterize the B. licheniformis YdaP, originally annotated in the genome database to be a pyruvate decarboxylase, and would have been the first representative from a thermotolerant organism. Here, we report on the thermostable activity of YdaP, as a pyruvate oxidase. We show that this enzyme has significant activity at 50°C to produce acetate from pyruvate.
Materials and Methods
Materials. All biochemical reagents used in this study were purchased from Sigma Aldrich or Fluka, Germany.
Isolation of B. licheniformis 9A. B. licheniformis 9A was isolated from soil collected from the Karoo region (Northern Cape Province, South Africa). Unless otherwise stated, B. licheniformis 9A was grown aerobically at 50ºC for 18 hours in 50 ml Tryptone Soya Broth (TSB) supplemented with glucose (0.25%) as a carbon source, with shaking at 150 rpm. The isolate was identified by analyzing the 16S rRNA gene sequence generated with species specific primers from genomic DNA isolated as described previously (Miller et al., 1999). The 16S rRNA PCR product was cloned into the pTZ57R/T vector (Fermentas Life Science) and transformed into E. coli Genehogs (Novagen) according to the manufacturer’s instructions.
Cloning and Overexpression of the B. licheniformis 9A YdaP. Primers specific to the annotated ydaP gene (Accession number CBE70488) were designed: YdaP-F (5’-ATGGATGGCAAACAAAACCGCAG-3’) and YdaP-R (5’-TAGGCGATCTCCCTTGGCATCATAC-3’) incorporating the NcoI and BamHI restriction sites (underlined). The PCR thermocycling conditions were set as 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 57°C for 4 min and 72°C for 2 min, and the extension step of 72°C for 1 min. The PCR product was cloned into pET-28a (Novagen), and transformed into E. coli Genehogs. The 1719 bp ydaP gene was amplified from genomic DNA cloned into the vector. The inserts from a selection of transformants were sequenced to identify a clone without PCR errors, which was subsequently transformed into E. coli Rosetta2 pLysS (Novagen) for overexpression of YdaP. Overexpression was conducted in an auto-induction medium (Grabski et al., 2005), containing Kanamycin (25 mg/ml) and chloramphenicol (25 mg/ml), and incubated aerobically for 16–24 hours at 37ºC. Cells were harvested by centrifugation at 10 000 × g and lysed with the Bugbuster extraction reagent (Novagen), supplemented with a Benzonnase nuclease (Novagen). The cellular debris was removed by centrifugation at 10 000 × g for 15 min at 4°C, and the crude extract was used for YdaP purification.
Purification of recombinant YdaP protein. Proteins in the crude extract were precipitated with 30% saturated ammonium sulphate, and incubated on ice for one hour. Following 0.22 µM filtration, the supernatant was used for hydrophobic interaction chromatography on a HiLoadTM Phenyl Sepharose XK16 column. The unbound proteins were eluted in buffer A (50 mM potassium phosphate, 1 M ammonium sulfate pH 5.8). The bound protein was eluted with a linear gradient of decreasing salt concentration (1 M ammonium sulfate in 25 mM potassium phosphate buffer (pH 5.8) at a flow rate of 2 ml min–1. Fractions showing activity where loaded onto a HiLoadTM 26/10 Q-Sepharose XK26 ion exchange column that was pre-equilibrated with 25 mM potassium phosphate buffer pH 5.8 (Buffer B). The unbound proteins were eluted in 5 column volumes of Buffer B. The bound protein was eluted using a linear gradient of increasing salt concentration from 0 to 1 M sodium chloride in 25 mM potassium phosphate buffer (pH 5.8) at a flow rate of 2 ml min–1. Fractions were collected, pooled, concentrated and tested for activity, and stored at –20°C.
Zymogram assay. YdaP activity was detected by staining the non-denaturing PAGE gel electrophoresis using a staining solution consisting of pyruvate as a substrate with TPP and MgCl2 dissolved in 0.1 M potassium phosphate buffer (pH 5.8), and 2,6-dichlorophenolindophenol (DCPIP) as an electron acceptor. The molecular weight of the active YdaP protein was estimated by using 10% Native-PAGE electrophoregram.
Pyruvate oxidase enzyme assays. The purified YdaP enzyme was assayed for pyruvate oxidase activity (EC 1.2.5.1) based on the reduction of 2,6-dichlorophenolindophenol (DCPIP) [20]. The reaction mixtures contained 250 mM potassium phosphate buffer (pH 5.8), 1mM TPP, 2 M sodium pyruvic acid, 100 mM MgCl2 and 1% Triton X-100 (v/v). The control contained purified enzyme and pyruvate was substituted with dH2O. After the reaction mixtures were prepared, 100 μl of the purified enzyme was added and incubated at 25ºC for 20 min, followed by addition of 100 μl of 300 µM 2,6-dichlorophenolindophenol (DCPIP). Changes in absorbance were monitored spectrophotometrically at 450 nm. The enzymatic activity was calculated using ε=0.218 mM–1 cm–1 (Cunningham & Hager, 1971; Blake & Hager, 1978). One unit (1U) of the YdaP activity was defined as the amount of YdaP catalyzing the production of 1 µM of acetate per min from pyruvate at 25°C. The activity of the YdaP enzyme was determined in 100 mM sodium phosphate/citrate buffers with pH values ranging from 4.6 to 6.0 and 100 mM potassium phosphate buffers in the pH range of 5.8 to 7.4, at 22°C. The rates of reaction were monitored and the absorbance was measured at OD450nm in 1 cm path length cuvettes. Thermostability assay was conducted by incubating YdaP preparations at a range of 30 to 80°C in 50 mM potassium phosphate buffer (pH 5.8) and 1% Triton X-100 was added for enzyme activation for 20 min, and the percentage residual activity was recorded. Various α-keto acid substrates were tested, including sodium pyruvate, α-ketobutyric acid, 3-methyl-2-oxopentanoic acid, indole-3-pyruvic acid, 4-hydroxyphenylpyruvic acid and phenylglyoxylic acid. The kinetic constants were determined using the direct linear plot of Eisenthal and Cornish-Bowden (1974), assayed at 50°C in 100 mM potassium phosphate buffer at the pH of 5.8.
HPLC Analysis of acetate production. The assay mixture (1 ml) contained 40 mM potassium phosphate buffer (pH 5.8), 10 mM MgCl2, 10 µM TPP, 10 mM sodium pyruvate and purified YdaP enzyme, and was incubated at 25°C for 20 min. The reaction mixture was centrifuged for 5 min at 16 200 × g before HPLC analysis. Reaction products were separated on a Rezex RHM monosaccharide H+ (8%) column (Phenomenex, USA) using 5 mM sulfuric acid as the mobile phase (Okuyama et al., 1989). The flow rate was set at 0.5 ml/min; the column oven temperature was 40ºC and pressure was set at 32 bars with an injection volume of 20 µl. The compounds were detected using RI and UV detectors, set to 215 nm, and compared to standards.
Nucleotide sequence analysis. DNA sequences were analysed using DNAMAN (Lynnon BioSoft) and Bio-Edit version 5.0.9 software (Hall, 1999). Comparison searches were performed using the gapped-BLAST program at the National Centre for Biotechnology Information (Altschul et al., 1997). Multiple sequence alignments were performed using ClustalW. The YdaP molecular weight was determined using DNAMAN and pI was predicted using the EXPASY website (www.expasy.org/). The B. licheniformis 9A ydaP gene sequence was deposited in the GenBank database under accession number CBE70488.
Homology modeling. The threading programs FUGUE (Shi et al., 2001) and MODELLER 9v4 (Sali, 1995) were used to generate structural YdaP models, using the L. plantarum POX (PDB 2EZ9) as a template. Stereochemical analysis of the YdaP structure was carried out using RAMPAGE (Lovell et al., 2001).
Results
The 16S rRNA sequence analysis showed 97% identity to B. licheniformis, designated isolate 9A in this study (Table 1). Sequence analysis revealed 1537/1569 bp differences compared to the ydaP gene sequence for B. licheniformis ATCC 14580 and contained a low mol% G+C content (49%).
Table 1. Blast (nucleotides) search results for the Bacillus 9A isolate 16S rRNA gene.
|
Strains
|
Accession number
|
Identity (%)
|
Evalue
|
|
B. licheniformis ATCC 14580
|
CP000002
|
1537/1569 (97%)
|
0.0
|
|
B. licheniformis DSM 13
|
AE017333
|
1537/1569 (97%)
|
0.0
|
|
B. licheniformis strain N8
|
DQ350834
|
1518/1551 (97%)
|
0.0
|
|
B. licheniformis
|
X684116
|
1520/1554 (97%)
|
0.0
|
|
B. licheniformis strain MML2501
|
EU344793
|
1517/1551 (97%)
|
0.0
|
The gene sequence encoded a putative protein of 573 amino acids with an estimated molecular weight of approximately 63 kDa, with a pI value of 5.83. The YdaP protein had shown identity to thiamine-dependent family proteins, containing the signature motif at amino acid positions G431-N459 (Fig. 1). A high level of identity was also seen with the uncharacterized ydaP genes from many other Bacillus organisms (B. subtilis NP 388315, 75%; B. pumilus YP 0014855662, 73%; B. amyloliquefaciens YP 00420086, 72%; and B. cereus ZP 03232334, 54%). Although only 75% sequence identity was observed with the EC 1.2.3.3 pyruvate oxidases, all of the Bacillus ydaP sequences clustered with this group of enzymes (Fig. 2).
Figure 1. Multiple sequence alignments of B. licheniformis YdaP with pyruvate oxidase from S. aureus, pyruvate oxidase from C. glutamicum, PDH from E. coli, L. plantarum and A. viridans.
Catalytic residues are indicated with the black boxes, the ThDP binding site is indicated with the red box and the substrate binding sites are indicated with the yellow boxes. Residues indicated with the green boxes are highly conserved, and thought to be involved in catalysis.
Figure 2. Phylogenetic alignment of B. licheniformis 9A YdaP in relation to uncharacterized YdaP sequences identified in other Bacillus sequences from the GenBank database, and representatives of the EC 1.2.3.3 and EC 1.2.2.2 type POX enzymes.
The expression of B. licheniformis YdaP using the pET system enables rigorous control of gene expression. A high level of YdaP expression was achieved under optimized conditions (San et al., 1994). Crude cell extracts were analysed by SDS-PAGE (Fig. 3). A protein band was evident at approximately 63 kDa, corresponding to the predicted molecular weight of B. licheniformis YdaP.
Figure 3. 12% SDS-PAGE gel electrophoresis showing the expression of YdaP with pET-28a (+) expression vector.
Lane M: Molecular weight marker; Lane 1: pET28a in E. coli; Lane 2: soluble fraction; Lane 3: soluble fraction; Lane 4: soluble fraction; Lane 5: soluble fraction treated at 65ºC; Lane 6: Ammonium sulfate treatment.
The purified YdaP enzyme showed a distinct band on SDS-PAGE. The subunit molecular weight was calculated to be approximately 63 kDa (Fig. 3). The YdaP protein showed identity to thiamine-dependent family proteins containing the signature motif at amino acid positions G431- N459 (Hawkins et al., 1989) (Fig. 1). This finding is in complete agreement with previous studies of other POX proteins (Patton et al., 2005, Schreiner & Eikmanns, 2005). Crude extracts of B. licheniformis YdaP (with a specific activity of 0.1 U/mg–1) were subjected to ammonium sulfate precipitation. This method does not result in detrimental effects on the enzyme activity and was employed to facilitate the binding of the crude enzyme to the hydrophobic interaction chromatography matrix. A 30% (w/v) ammonium sulfate concentration was used and resulted in an increase in YdaP specific activity to 2.62 U/mg–1 (Table 2). Following hydrophobic interaction chromatography, ion exchange chromatography was used to further purify the active B. licheniformis YdaP protein. This strategy achieved the highest purification level (Zhang & Hager, 1987) of the YdaP enzyme, as determined by SDS-PAGE (Fig. 3). Fractions exhibiting the YdaP protein activity were collected, pooled and concentrated for further characterization.
Table 2. Summary of the YdaP purification steps.
|
Sample
|
Volume (ml)
|
Total Protein (mg)
|
Total Activity (U)
|
Specific Activity (U/mg)
|
Yield (%)
|
Purification (fold)
|
|
Crude extract
|
15
|
795
|
303
|
0.1
|
100
|
1
|
|
(NH4)2SO4 precipitation
|
15
|
138
|
67.1
|
2.62
|
17
|
26.2
|
|
Phenyl-Sepharose
|
15
|
116
|
54
|
4.86
|
15
|
48.6
|
Figure 4. (A) Q-Sepharose ion exchange chromatography of B. licheniformis YdaP (B) 10% SDS-PAGE electrophoregram;
Lane M: Molecular weight marker; Lane 1: Flow-through fraction of YdaP protein; Lane 2: First fraction of YdaP soluble protein; Lane 4–9: Second fraction of the soluble of putative YdaP protein.
The molecular weight of the active YdaP protein was estimated to be approximately 250 kDa, which represent the active form of the YdaP enzyme (Fig. 5). This observation implies that the YdaP protein is a homotratermer. YdaP activity was detected by staining the non-denaturing PAGE gel electrophoresis using a staining solution consisting of pyruvate as a substrate, with TPP and MgCl2 dissolved in 0.1 M potassium phosphate buffer (pH 5.8), and 2,6-dichlorophenolindophenol (DCPIP) as an electron acceptor. The reaction resulted in the formation of a white precipitate in the gel (Fig. 4), and this observation confirmed that the stained band corresponded to the B. licheniformis YdaP.
Figure 5. 10% Native-PAGE electrophoregram showing the YdaP activity;
Lane M: Molecular marker; Lane 1: E. coli/pET28a vector; Lane 2: Crude cell extracts of YdaP enzyme; Lane 3: Semi-purified YdaP enzyme (HIC fraction).
The pH activity-profile of B. licheniformis YdaP was performed over the pH range of 4.6 to 7.4 (Fig. 4.10). YdaP showed maximum activity at pH 5.8. This pH optimum is slightly lower than values reported for the E. coli, L. plantarum (Lorquet et al., 2004) and A. viridans POX (Juan et al., 2007), all of which show optimal values at pH 6.0. The B. licheniformis YdaP retained approximately 50% activity at pH values of 5.4 and 6.5, and had detectable activity in the range from pH 4.6 to 7.4.
The enzyme was found to perform optimally at 50ºC (Table 3) and pH 5.8 (Fig. 6). The activity was stable at 50°C, but was rapidly inactivated at higher temperatures, retaining only 11% of initial activity at 80°C (Fig. 6).
YdaP activity was tested on a range of different substrates (Table 4). The highest activity was achieved with pyruvate (C3), and activity decreased as the substrate chain length increased (Table 4). Low activity was detected with aromatic and larger branched chain substrates.
Table 3. Effect of temperature on YdaP stability
|
Temperature (°C)
|
Relative activity (%) ± S.D.
|
|
30
|
86±4.0
|
|
40
|
87±4.3
|
|
50
|
100±4.4
|
|
60
|
82±4.9
|
|
70
|
13±1.1
|
|
80
|
11±1.1
|
Table 4. Comparison of the substrate specificities of the YdaP enzyme of B. licheniformis 9A strain
|
Substrate
|
Carbon number
|
Structure
|
Relative activity (%)
|
|
Pyruvic acid
|
C3
|
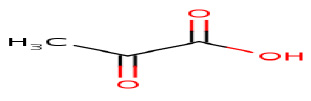
|
100±0.0
|
|
α-Ketobutyric acid
|
C4
|
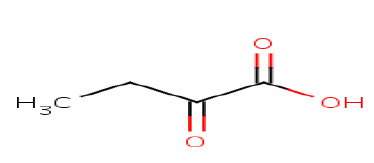
|
23±3.1
|
|
3-Methyl-2-oxopentanoic acid
|
C6
|
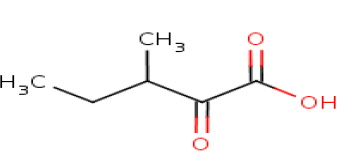
|
18±2.3
|
|
Phenylglyoxylic acid
|
C8
|
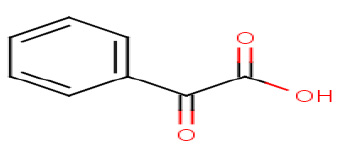
|
12±0.6
|
|
4-Hydroxyphenylpyruvic acid
|
C9
|
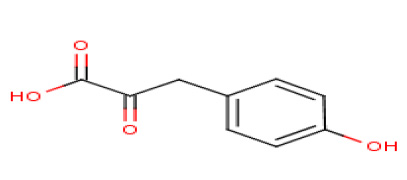
|
12±0.9
|
|
Indole-3-pyruvic acid
|
C11
|
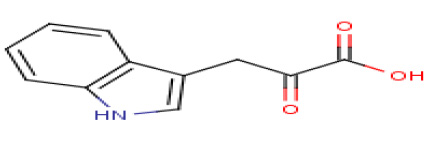
|
11±2.4
|
Figure 6. pH profile of the recombinant YdaP based on relative activity at 50°C and at different pH values. YdaP showed maximum activity at pH 5.8.
Kinetic constants were estimated as averages of values calculated from a direct linear plot. The purified recombinant YdaP displayed similar substrate affinities and rates of reaction to both, the crude extract and partially purified enzyme. The recombinant enzyme had a KM of 4.4 mM for pyruvate and a Vmax of 4.86 U/mg protein, and Kcat of 25.3s–1 (Table 5) when assayed at a temperature of 50ºC and a pH of 5.8. The recombinant enzyme displayed a higher affinity for pyruvate than C. glutamicum POX (30 mM) (Schreiner & Eikmanns, 2005).
Table 5. The kinetic data of the partially purified YdaP on pyruvate substrate
|
Sample
|
Km (mM)
|
Vmax(U/mg)
|
kcat (s–1)
|
kcat/Km (s–1 mM–1)
|
|
Phenyl-Sepharos (HIC)
|
4.4
|
4.86
|
25.3
|
11.5
|
For the HPLC analysis of B. licheniformis YdaP enzyme, pyruvate and acetate were selected as standards for quantification using the Rexez RHM H+ column. Various HPLC conditions were tested to achieve the best resolution of acetate as a standard. The retention time of acetate was 15.44 min. A single product peak was detected using pyruvate as substrate with B. licheniformis YdaP preparations, corresponded exactly to the acetate peak generated with the standard. Acetate was detected in all YdaP reactions at pyruvate substrate concentrations ranging from 10 mM to 200 mM. However, in all reactions, the estimated acetate yield was approximately 1 mM; that is, the product yield did not correlate with the substrate concentration.
The YdaP model was generated using the L. plantarum POX (PDB 2EZ9) as a template, which shares 35% sequence identity to YdaP. The model of the YdaP monomer (Fig. 7) is compatible with the α/β topology observed in all of the crystallized TPP-dependant decarboxylases (Wille et al., 2006, Juan et al., 2007), comprising the N-terminal Pyruvate domain, FAD domain and the C-terminal ThDP domain.
Figure 7. Cartoon representation of the Bacillus licheniformis 9A YdaP monomer, showing the three-domain structure of the YdaP subunit.
Discussion
Phylogenetic analysis using closely related 16S rRNA gene sequences from other bacterial species had shown that phylogenetically, the 9A isolate was very closely related to other Bacillus species. The species of licheniformis, amyloliquefaciens (Priest et al., 1987), subtilis (Priest et al., 1988) and cereus (Radnedge et al., 2003) were closest to the 9A isolate (Fig. 2). GenBank analysis and alignment studies revealed that the B. licheniformis 9A strain ydaP gene was encoding a pyruvate oxidase (POX; EC 1.2.5.1). The significantly high sequence identity between homologous genes of these species suggested that they were closely related; they encoded an uncharacterized pyruvate oxidase. Sequence alignment analysis revealed that this group of enzymes belongs to the family of ThDP dependent enzymes responsible for the decarboxylation of pyruvate. B. licheniformis YdaP shared many biochemical properties with other pyruvate oxidases, including C. glutamicum POX (Patton et al., 2005; Schreiner & Eikmanns, 2005), other than its thermostability. The B. licheniformis YdaP enzyme represents a new subfamily of pyruvate oxidases. This result was based on the phylogenetic tree as the pyruvate oxidases group together under the same cluster, however, the YdaP enzyme is forming a new clade with the Bacillus genera (Fig. 2).
The homology analysis of amino acids deduced from the ydaP gene sequence demonstrates much less identity with the acetyl-phosphate and H2O2 forming pyruvate oxidases (POX; EC 1.2.3.3) of L. plantarum (35%). In contrast, the fact that the B. licheniformis YdaP enzyme reaction is phosphate independent and that the product of the B. licheniformis YdaP enzyme reaction is acetate instead of acetyl-phosphate argue against a close relationship between the B. licheniformis YdaP enzyme and the lactobacterial and E. coli pyruvate oxidases.
Comparative analysis of the complete amino acid sequence of B. licheniformis YdaP using PCR specific primers based on the ydaP gene sequence, aligned with S. aureus, C. glutamicum, E. coli, L. plantarum and A. viridans (EC 1.2.3.3), provided some information based on their ThDP domains and is high enough to directly infer into their evolutionary relationships. Overall phylogeny analysis of these pyruvate oxidase (EC 1.2.5.1) sequences had shown them to group together, forming a single cluster. Based on their sequence divergence, the apparently observed phylogenetic cluster of this group was subsequently divided into two distinct clades and was supported by a bootstrap value of 84%.
The B. licheniformis YdaP enzyme was purified from cell extracts and most of the activity was found in the soluble cytoplasmic fraction. The enzyme was shown to be activated by the Triton X-100 detergent. A variety of the membrane associated enzymes have been shown to require detergents for their catalytic activity, with the pyruvate oxidase from E. coli among them (Blake, 1982). The YdaP has unique characteristics differentiating it from previously characterized EC 1.2.5.1 pyruvate oxidases (Neale et al., 1987), in that it displays thermostability and catalyzes reactions with a broad range of substrates including linear, branched and aromatic oxo acids. As for the other pyruvate oxidases (Schreiner & Eikmanns, 2005), YdaP displays Michaelis-Menten kinetics with pyruvate as substrate, however, YdaP showed higher affinity for pyruvate (Km=4.4 mM) when compared to that of C. glutamicum (Km=30 mM) (Schreiner & Eikmanns, 2005). This enzyme is a homotetrameric flavoproteins consisting of 63 kDa subunits with tightly bound FAD, and it requires TPP and a divalent metal cation such as Mg2+, Mn2+ and Co2+ for enzymatic activity.
Biochemical characterization of the B. licheniformis 9A YdaP indicated that it was a pyruvate oxidase (EC 1.2.5.1), catalyzing the oxidative decarboxylation of pyruvate to generate acetate and carbon dioxide. Due to the high sequence identity to the annotated B. licheniformis genome YdaP (97%), we propose that the Bacillus ydaP genes encode a pyruvate oxidase, and not in fact a pyruvate decarboxylase. Refinement of the annotation has subsequently re-classified the ydaP gene as a pyruvate oxidase as we have shown. This illustrates the danger in assigning function based on the sequence data alone, but also how the bioinformatics tools have improved in recent years to generate more reliable annotations.
This study demonstrated that the B. licheniformis YdaP is more stable compared to pyruvate oxidases from other microorganisms (Neumann et al., 2008; Patton et al., 2005). The enzyme was stable at higher temperatures than other pyruvate oxidases, such as the E. coli POX (temperature-stability of 25oC and pH 6.0) (Mather & Gennis, 1985).
Investigations of the substrate specificity of the B. licheniformis YdaP enzyme showed a higher specific activity for pyruvate than for α-ketobutyric acid (23%), 3-methy-2-oxopentanoic (18%) and other substrates used in the study, suggesting a strong preference for shorter chain length substrates. The catalytic cavity of the B. licheniformis YdaP contains some bulky residues, such as histidine and tyrosine. The presence of bulky residues within the catalytic cavity may limit the entry of larger substrates into the substrate channel. A strong preference for pyruvate and other smaller α-keto acids substrates suggests that these may be the in vivo substrate, and the principal role of this enzyme could be the production of acetate.
The YdaP enzyme was purified to near homogeneity by ammonium sulfate precipitation, hydrophobic interaction and ion exchange chromatography. YdaP activity was initially detected by a zymogram, where a band of approximately 250 kDa formed a precipitate in the presence of pyruvate and DCPIP as a cofactor (Fig. 4). This suggested that the active form of the YdaP enzyme functions as a homotetramer, a feature common for the TPP family proteins. The production of acetate was confirmed by HPLC. HPLC analysis was used to validate the conversion of pyruvate by B. licheniformis YdaP to acetate. However, acetate was produced at a constant concentration of approximately 1mM, irrespective of the substrate concentration. Similar results were obtained in previous studies of acetate production, like the 0.96 mM reported for C. glutamicum POX (Schreiner & Eikmanns, 2005). It is likely that these results reflect a reaction limitation imposed by the concentration of the DCPIP electron acceptor (300 µM) added to the reaction. The stoichiometric reaction of DCPIP in the enzyme assay leads to speculate that the DCPIP could be limiting the completion of conversion of the substrate to acetate. Therefore, the obtained result suggests that the increasing of the DCPIP concentration might increase the acetate product. The presented study describes and validates a HPLC method for determination of acetate as a method of choice. It is likely that HPLC has the potential to rapidly identify and quantify acetate, and could be a significant contributor in development and screening of acetate products within a family of pyruvate oxidases.
The 3 Dimension structural model of YdaP of B. licheniformis was constructed based on the closest similarity to the experimentally determined structure of LpPOX (Wille et al., 2006). Despite the overall identities between the primary structures of the YdaP enzyme from B. licheniformis and the L. plantarum pyruvate oxidase, the alignment studies revealed some common features in the L. plantarum pyruvate oxidase. The overall topology of the B. licheniformis YdaP was similar to the known pyruvate oxidase crystal structures (Neumann et al., 2008). The structural model generated was assessed and revealed to be in a good agreement with template structure suggesting the accuracy spectrum of the B. licheniformis YdaP model.
The structure of the ThDP motif was identical to that found in the other pyruvate oxidases and the residues (Asp313 and Ala314) (Muller & Schulz, 1993; Muller et al., 1994), suggested to be involved in metal ion cofactor binding (MgCl2) and also showed positional conservation (Neumann et al., 2008).
YdaP showed a preference for short chain length substrates, suggesting that the dimensions of the active site might be relatively small. A comparison of the YdaP active site binding pocket with that of other pyruvate oxidases (Muller et al., 1994) showed that bulky hydrophobic amino acid residues Tyr469, His476 and Tyr479 formed part of the active site cavity in YdaP. In L. plantarum POX, these correspond to amino acid residues Trp479, Ile480 and Glu483. These residues are presumably critical for the catalytic activity of pyruvate oxidases, and have been proposed to be involved in substrate binding. This observation suggested that these residues would negatively influence the accessibility of large substrates (e.g., aromatic) into the catalytic centre, thereby restricting the activity of YdaP for short chain substrates. This information may assist in studies aimed at engineering the catalytic active site of the enzyme to improve accessibility of larger substrates into the active site.
Acknowledgement
The authors gratefully acknowledge TMO Renewable and the National Research Foundation for the financial support of this project.
References
Altschul SF, Madden TL, Schaffer AA, Zang JZ, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. doi: 10.1093/nar/25.17.3389
Bertagnolli BL, Hager LP (1991) Activation of Escherichia coli pyruvate oxidase enhances the oxidation of hydroxyethylthiamin pyrophosphate. J Biol Chem 266: 10168–10173
Blake R, O’Brien TA, Gennis RB, Hager LP (1982) Role of the divalent metal cation in the pyruvate oxidase reaction. J Biol Chem 257: 9605–9611
Blake R, Hager LP (1978) Activation of pyruvate oxidase by monomeric and micellar amphiphiles. J Biol Chem 253: 1963–1971
Chang YY, Cronan JE, Jr (1986) Molecular cloning, DNA sequencing, and enzymatic analyses of two Escherichia coli pyruvate oxidase mutants defective in activation by lipids. J Bacteriol 167: 312–318
Chang YY, Cronan JE, Jr (1988) Common ancestor of Escherichia coli pyruvate oxidase and the acetohydroxy acid synthases of the branched-chain amino acid biosynthetic pathway. J Bacteriol 170: 3937–3945
Chang YY, Cronan JE, Jr (1995) Detection by site-specific disulfide cross-linking of a conformational change in binding of Escherschia coli pyruvate oxidase to lipid bilayers. J Biol Chem 270: 7896–7901
Claus SC, Berkeley RCW (1986) Genus Bacillus cohn 1872. In Sneath PHA et al., eds. pp 1105–1139. Baltimore, MD: Williams and Wilkins Co
Cunningham CC, Hager LP (1971) Crystalline pyruvate oxidase from Escherichia coli phospholipids as an allosteric effector for the enzyme. J Biol Chem 264: 1583–1589
Duggleby RG (2006) Domain relationships in thiamine diphosphate-dependent enzymes. Acc Chem Res 39: 550–557. doi: 10.1021/ar0680222
Diallo MD, Martens M, Vloemans N, Cousin A, Vandekerckhov TTM, Neyra M, de Lajudie P, Willems A, Gillis M, Vyverman W, Van der Gucht K (2004) Phylogenetic analysis of partial bacterial 16S rDNA sequence of tropical grass pasture soil under Acacia tortilis subsp raddiana in Senegal. Syst App Microbiol 27: 238–252. doi: 10.1078/072320204322881862
Erickson RJ (1976) Industrial application of the Bacilli: A review and prospectus. In Schlensiger D ed, pp 406–419. Microbiology. Washington, DC: Amer Soc Microbiol
Eveleigh DE (1981) The microbial production of industrial chemicals. Scientific American 245: 155–178
Goffin P, Muscariello L, Lorquet F, Stukkens A, Prozzi D, Sacco M, Kleerebezem M, Hols P (2006) Involvement of pyruvate oxidase activity and acetate production in the survival of Lactobacillus plantarum during the stationary phase of aerobic growth. Appl Environ Microbiol 72: 7933–7940. doi: 10.1128/AEM.00659-06
Grabski A, Mehler M, Drott D (2005) The overnight expression autoinduction system: High-density cell growth and protein expression while you sleep. Nature Meths 2: 233–235. doi: 10.1038/nmeth0305-233
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 95–98
Hamilton SE (1986) Identification of the high-affinity lipid binding site in Escherichia coli pyruvate oxidase. Biochemistry 25: 8178–8183. PMID: 3545288
Hawkins CF, Borger A, Perham RN (1989) A common structural motif in thiamin pyrophosphate-binding enzymes. FEMS Lett 255: 77–82.
Juan ECM, Hoque MM, Hossain MT, Yamamoto T, Imamura S, Suzuki K, Sekiguchi T, Takénaka A (2007) The structures of pyruvate oxidase from Aerococcus viridans with cofactors and with a reaction intermediate reveal the flexibility of the active-site tunnel for catalysis. Acta Crystallogr Sect F Struct Biol Cryst Commun 63: 900–907. doi: 10.1107/s1744309107041012
Lorquet F, Goffin P, Muscariello L, Baudry JB, Ladero V, Sacco M, Kleerebezem M, Hols P (2004) Characterization and functional analysis of the poxB gene, which encodes pyruvate oxidase in Lactobacillus plantarum. J Bacteriol 186: 3749–3759. doi: 10.1128/JB.186.12.3749-3759-2004
Lovell SC, Davis JW, Arendall WB, 3rd de Bakker PI, Word JM, Richardson MG, Richardson JS (2001) Structure validation by Calpha geometry phi, psi and C beta deviation. Proteins 50: 43–450. PMID: 12557186
Mather M, Schopfer LM, Massey V, Gennis RB (1982) Studies of the flavin adenine dinucleotide binding region in Escherichia coli pyruvate oxidase. J Biol Chem 257: 12887–12892
Miller DN, Bryant JE, Madsen EL, Ghiorse WC (1999) Evaluation and optimization of DNA extraction and purification procedures for soil sediment samples. Appl Environ Microbiol 65: 4715–4724. PMID: 10543776. PMCID: PMC 91634
Muller YA, Schulz GE (1993) Structure of the thiamine- and flavin-dependent enzyme pyruvate oxidase. Science 259: 965–967
Muller YA, Schumaker G, Rudolph R, Schulz GE. (1994) The refined structure of stabilized mutant and of wild-type pyruvate oxidase from Lactobacillus plantarum. J Mol Biol 237: 315–335
Neale AD, Scopes RK, Wettenhall RE, Hoogenraad NJ (1987) Pyruvate decarboxylase of Zymomonas mobilis: isolation, properties and genetic expression in Escherichia coli. J Bacteriol 169: 1024–1028. PMCID: PMC211896
Neumann P, Weidner A, Pech A, Stubbs MT, Tittmann K (2008) Structural basis for membrane binding and catalytic activation of the peripheral membrane enzyme pyruvate oxidase from Escherichia coli. Proc Nat Acad Sci 105: 17390–17395. doi: 10.1073/pnas.080527105
O’Brien TA, Blake R, Gennis RB (1977) Regulation by lipids of cofactor binding to a peripheral membrane enzyme: Binding of thiamine pyrophosphate to pyruvate oxidase. Biochemistry 16: 3105–3109
Okuyama T, Takata M, Takahashi K (1989) High-performance liquid chromatographic analysis of naturally occurring glycosides and saponins. J Chromatogr 466: 390–398. doi: 10.1016/s0021-9673(01)84636-7
Patton TG, Rice KC, Foster MK, Bayles KW (2005) The Staphylococcus aureus cidC gene encodes a pyruvate oxidase that affects acetate metabolism and cell death in stationary phase. Mol Microbiol 56: 1664–1674. doi: 10.1111/j.1365.2005.04653.x
Priest FG, Goodfellow M, Shute LA, Berkeley RCW (1987) Bacillus amyloliquefaciens sp. Nov Rev Int J Syst Bacteriol 37: 69–71. doi: 10.1099/00207713-37-1-69
Priest FG, Goodfellow M, Todd C (1988) A numerical classification of the genus Bacillus. J Gen Microbiol 134: 1847–1882. doi: 1099/00221287-134-7-1847
Radnedge L, Agron PG, Hill KH, Jackson PJ, Ticknor LO, Keim P, Andersen GL (2003) Genome differences that distinguish Bacillus anthracis from Bacillus cereus and Bacillus thuringiensis. Appl Environ Microbiol 69: 2755–2764. doi: 10.1128/AEM.69.5.2755-2764.2003
Rey MW, Ramaiya P, Nelson BA, Brody-Karpin SD, Zaretsky EJ, Tang M, Lopez de Leon A, Xiang H, Gusti V, Clausen IG, Olsen PB, Rasmussen MD, Andersen JT, Jørgensen PL, Larsen TS, Sorokin A, Bolotin A, Lapidus A, Galleron N, Ehrlich SD, Berka RM (2004) Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparison with closely related Bacillus species. Genome Biol 5: R77. doi: 10.1186/gb-2004-5-10-r77
Šali A (1995) Modelling mutations and homologous proteins. Curr Opin Biotech 6: 437–451. doi: 10.1016/0958-1669(95)80074-3
San KY, Bennet GN, Aristidou AA, Chou CH (1994) Strategies in high-level expression of recombinant protein in Escherichia coli. Ann N Y Acad Sci 721: 257–267. doi: 10.1111/j.1749-6632.1994.tb47399.x
Schreiner ME, Eikmanns BJ (2005) Pyruvate: Quinone oxidoreductase from Corynebacterium glutamicum: purification and biochemical characterization. J Bacteriol 187: 862–871. doi: 10.1128/JB.187.3.862-871
Sedewitz B, Schleifer KH, Gotz F (1984) Purification and biochemical characterization of pyruvate oxidase from Lactobacillus plantarum. J Bacteriol 160: 273–278. PMCID: PMC214712.
Shi J, Blundell TL, Mizuguchi K (2001) Sequence structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 310: 243–257. doi: 10.1006/jmbi.2001.4762
Steele, H L (2008) Advance in recovery of novel biocatalysts from metagenomes. Mol Microbiol Biotechnol 16: 25–37. doi: 10.1159/000142892
Tomar A, Eiteman MA, Atman, E (2003) The effect of acetate pathway mutations on the production of pyruvate in Escherichia coli. Appl Microbiol Biotechnol 62: 76–82. doi: 10.1007/s00253-003-1234-6
Veith B, Herzberg C, Steckel S, et al., (2004) The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol 7: 204–211. doi: 10.1159/000079829
Wang A. Y, Chang YY, Cronan, JE (1991) Role of the tetrameric structure of Escherichia coli pyruvate oxidase in enzyme activation and lipid binding. J Biol Chem 266: 10959–10966
Wille G, Meyer D, Steinmetz A, Hinze E., Golbik R, Tittmann K (2006) The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat Chem Biol 2: 324–328. doi: 10.1038/nchembio.788
Yilmaztekin M, Erten H, Cabaroglu T (2008) Production of isoamyl acetate from sugar beet molasses by Williopsis saturnus var. saturnus. J Inst Brewing 114: 34–38. doi: 10.1016/j.foodchem.2008.05.079
Yoneda N, Kusano S, Yasui M, Pujado P, Wilcher S (2001) Recent advances in processes and catalysts for the production of acetic acid. Appl Catalysis 221: 253–265. doi: 10.1016/s0926-860x(01)00800-6
Zhang TF, Hager LP (1987) A single-step large-scale purification of pyruvate oxidase. Arch Biochem Biophys 257: 485–487. PMID: 3310893